PMCF, as part of the Post-Market Surveillance
Applicable since May 26, 2020, the new Medical Devices Regulation (MDR) implies new obligations for manufacturers to ensure the safety and efficacy of medical devices. Among them, manufacturers must set up a Post-Market Surveillance system (PMS) for each device placed on the market. This must be documented and continuously updated over time, considering the risk class and type of device concerned.
The PMS was presented in MDR 2017/745 in Article 2, definition 60: “’Post Market surveillance’ means all activities carried out by manufacturers in cooperation with other economic operators to institute and keep up to date a systematic procedure to proactively collect and review experience gained from devices they place on the market, make available on the market or put into service for the purpose of identifying any need to immediately apply any necessary corrective or preventive actions”.
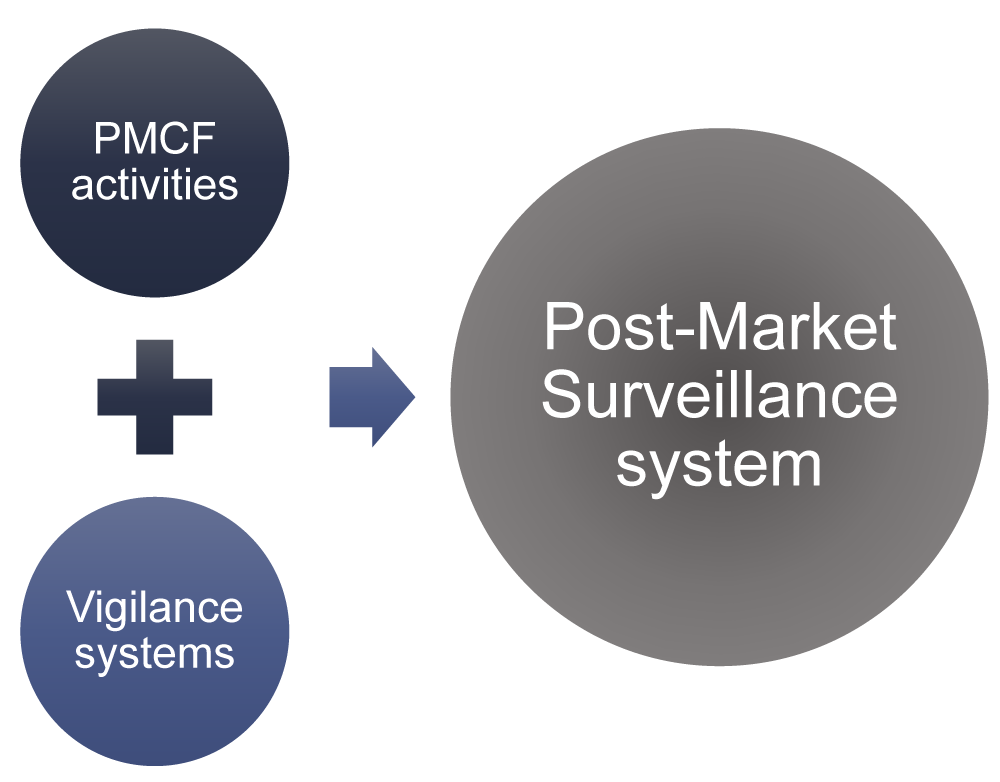
The Post-Market Clinical Follow-up (PMCF) is included in the PMS, in addition to the vigilance systems. Both PMCF and vigilance are complementary components of a PMS system, which describes a comprehensive strategy for monitoring the post-market safety and performance of medical devices.
PMS is a broad system that encompasses both Vigilance and PMCF
What is the Post-Market Clinical Follow-up (PMCF)?
The PMCF (Post-Market Clinical Follow-up) is performed after a medical device has been placed on the market. The goal of the PMCF is to be able to update the clinical evaluation report with new data on patient safety and device performance following the CE marking of the device.
The Annex XIV Part B of the MDR is entirely dedicated to the PMCF and defines it as: “PMCF shall be understood to be a continuous process that updates the clinical evaluation referred to in Article 61 and Part A of this Annex and shall be addressed in the manufacturer’s post-market surveillance plan”.
Annex XIV Part B of the MDR

The overall PMCF activities should be documented in a PMCF plan and the results compiled in a PMCF report that is part of the Clinical Evaluation Report (CER) of the device.
Definitions of PMS plan / PMCF plan / PMCF report / CER
PMS
Plan
• Details the strategy for continously monitoring and collecting information avout safety and performance of a device throughout its entire lifetime
• Incorporates PMCF activities and vigilance systems
• Is part of the manufacturer’s quality system
PMCF
Plan
• Outlines the manufacturer’s planned procedures and methods for collecting clinical data for post-market follow-up
• Complements the data obtained during the pre-market phase
PMCF
Report
• Is produced to provide the results obtained during PMCF activities
• Is fully integrated as part of the technical documentation and more specifically, it’s outcome included in the cilinical evaluation report
Clinical Evaluation Report (CER)
• Gathers the clinical evidence and documents the conclusions of a clinical evaluation of a medical device
• Is an essential step in obtaining CE marking of a device and must be regulary updated during the PMS
What are the common PMCF activities that exist?
There are many different methods to collect data for PMCF purposes. The choice of one or more PMCF activities depends on the objective, the evidence gaps to be filled, and the device risk class and type.
Whatever methods are used, they must be justified and their designs appropriate for the purpose. This is key to the successful implementation of a good PMCF that will comply with the requirements of the MDR and to the admissibility of the dossier by the notified bodies.
The most common PMCF activities:
Literature and published research
Screaning of scientific literature to collect information on clinical performance and safety on the device.
Third-party registries
Collecting data in registries which are databases established by private or public organizations for various reasons.
Investigator-initiated studies (ISS)
Collecting data on an ongoing basis via investigators, which manufacturers sponsors. In return, manufacturers have access to use the data for regulatory and marketing purposes.
PMCF survey
Collecting clinical data from users of medical devices (primarily healthcare professionnals, but also patients) through their responses to a series of pre-established questions in a survey.
PMCF case series
Collecting clinical data from activity that includes a series of cases or patients (years of data on the effects and use of medical devices).
PMCF studies
Collecting high quality of clinical data in interventional or observational studies which evaluate the medical device when used in accordance with its intended purpose and instructions for use.
What are PMCF studies?
PMCF studies are one of the options available for PMCF in post-market surveillance.
They are intended to answer specific questions regarding the clinical safety or performance of a device when used in accordance with its approved labeling.
When considering the overall benefits and risks of a device for marketing authorization, uncertainties may remain. To collect additional clinical data, PMCF studies should be conducted if residual risks are identified or if there is a need for clarification of long-term clinical performance that may impact the benefit/risk ratio of the device.
PMCF studies may also be indicated to address emerging concerns resulting from post marketing adverse event trends, information from the literature, signals from adverse event reports, active surveillance programs, or other sources.
Guidance on conducting PMCF studies during Market surveillance is provided in MEDDEV 2.12/2 REV.2.
How to perform a PMCF study under the MDR?
Specific requirements for designing, conducting and reporting clinical Investigations are documented in Annex XV of the MDR.
First, PMCF studies must comply with applicable laws and regulations, and follow relevant guidelines and standards.
The design and methodology of the PMCF study must be described in a Clinical Investigation Plan (CIP) or study protocol that will include a description of the rationale, objectives, endpoints, and statistical methods that must be adapted to achieve the stated objectives. The study must be carried out with adequate controls in place to be in full compliance with the CIP.
A Statistical Analysis Plan (SAP) is also required to provide a detailed description of the statistical methods. It is this document that will dictate how the analysis of the data is to be performed by a person with appropriate statistical expertise.
The final Study Report should describe the results of the analysis in detail and provide conclusions related to the intended objectives.
Quinten Medical Device PMCF studies
Our retrospective PMCF studies conducted on Real-World Data (RWD) are designed to produce Real World Evidence (RWE) about the safety and performance of devices in normal use.
We propose an innovative approach to generate robust clinical data and evidence that meet MDR requirements and match notified bodies expectations.
Ressources
– MDR : Regulation (EU) 2017/745 of the European parliament and of the council of 5 April 2017 on medical devices
– Guidance document Medical devices – Market surveillance – Post Market Clinical Follow-up studies – MEDDEV 2.12/2 rev.2
– MDCG 2020-13 Clinical evaluation assessment report template
– ISO 14155:2020 – Clinical investigation of medical devices for human subjects – Good clinical practice
– ICH guideline E6 on good clinical practice
To learn more about our offer towards PMCF studies, please visit the dedicated page